
Theory of Coarse-Graining and Multiscale Phenomena
The Voth group develops theoretical and computational frameworks to study multiscale phenomena in biology and materials science. We work on both new theoretically-informed conceptual models as well as more quantitative, systematic, bottom-up modeling methods. Since 2005, our group has been one of the leaders in the development of methods for defining coarse-grained (CG) interactions from atomistic simulations in the condensed phase. We have since contributed to systematic methods for defining optimal choices of coarse-graining mapping, observable representation, and effective interactions for large biomolecules. Also, we have developed new enhanced sampling methods, the ultra coarse-graining framework, and new approach to coarse-grained dynamics. We aim to research novel mapping schemes and reactive coarse-graining methods in the future. The development of these methods is tightly coupled to our work on molecular and reactive simulations.
The Theory of Multiscale Coarse-Graining (MS-CG)
The multiscale coarse-graining (MS-CG) methodology provides a systematic, bottom-up way to calculate effective CG interactions based on rigorous statistical mechanics. It seeks to approximate the many-body potential of mean force by variationally minimizing the difference between CG forces at the mapped fine-grained reference forces (a.k.a, “force matching”). This method is related to liquid state theory and the Yvon-Born-Green equation. The code implementing the MS-CG methodology is available for download here and is available as a fix in LAMMPS through the OpenMSCG package.
Recent methodological work on MS-CG has included the inclusion of three-body interactions as well as formulations for both the constant NVT and constant NPT ensembles. We have also explored center-of-charge mappings as an alternative to the more traditional center-of-mass and center-of-geometry mappings. MS-CG models have been applied to wide range of systems including common solvents (e.g., methanol, hexane, water, ionic liquids), membrane systems, carbohydrates, polyglutamine aggregation, peptide secondary structures, and larger proteins.
Using MS-CG, the sensitivity of coarse-grained models to changes in the underlying FG model from which it was derived can be studied. This method provides a low-noise, computationally efficient way to calculate this sensitivity. The sensitivity can be used to accurately calculate alchemical transferability across interaction parameters to first order at low computational cost. Additionally, we used MS-CG to investigate the challenges of representing observables in CG models. We present a condition that can ensure that a CG observable reproduces the distribution of the FG observable projected onto CG model.
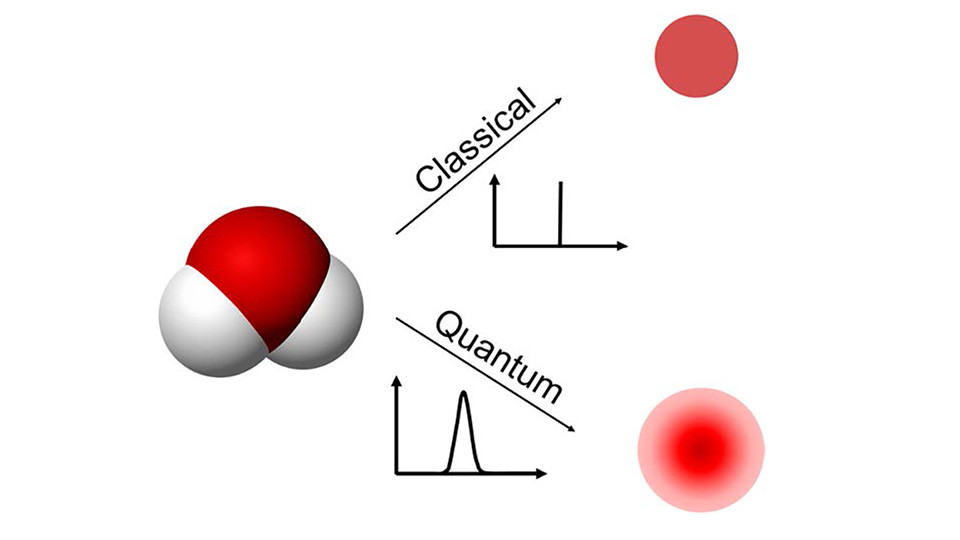
Recently, we extended the MS-CG methodology by explicitly including the order parameter dependent interactions to better reproduce the structural correlations at the inhomogeneous system. Another extension was to develop a theoretical methodology of coarse-graining for the quantum mechanical regime based on the MS-CG methodology. For the quantum MS-CG (qMS-CG) method, the Feynman path integral description of quantum statistical mechanics was combined with the variational force-matching method.
Relevant Papers:
-
Han, Y., Jin, J., Wagner, J. W., Voth, G. A. (2018). Quantum Theory of Multiscale Coarse-Graining. Journal of Chemical Physics, 148, 102335. doi: 10.1063/1.5010270
-
Wagner, J. W., Dannenhoffer-Lafage, T., Jin, J., Voth, G. A. (2017). Extending the Range and Physical Accuracy of Coarse-Grained Models: Order Parameter Dependent Interactions. Journal of Chemical Physics, 147, 044113. doi: 10.1063/1.4995946
-
Wagner, J. W., Dama, J. F., Voth, G. A. (2016). On the representability problem and the physical meaning of coarse-grained models. Journal of Chemical Physics, 145, 044108. doi: 10.1063/1.4959168
-
Wagner, J. W., Dama, J. F., Voth, G. A. (2015). Predicting the Sensitivity of Multiscale Coarse-Grained Models to their Underlying Fine-Grained Model Parameters. Journal of Chemical Theory and Computation, 11(8), 3547-3560. doi: 10.1021/acs.jctc.5b00180
-
Cao, Z., Voth, G. A. (2015). The Multiscale Coarse-Graining Method. XI. Accurate Interactions Based on the Centers of Charge of Coarse-Grained Sites. Journal of Chemical Physics. 143, 243116. doi: 10.1063/1.4933249
-
Noid, W. G., Liu, P., Wang, Y., Chu, J.-W., Ayton, G. S., Izvekov, S., Andersen, H. C., Voth, G. A., (2008). The multiscale coarse-graining method. II. Numerical implementation for coarse-grained molecular models. Journal of Chemical Physics, 128, 244115. doi: 10.1063/1.2938857
-
Noid, W. G., Chu, J.-W., Ayton, G. S., Krishna, V., Izvekov, S., Voth, G. A., Das, A., & Andersen, H. C. (2008). The multiscale coarse-graining method. I. A rigorous bridge between atomistic and coarse-grained models. Journal of Chemical Physics, 128(24), 244114. doi: 10.1063/1.2938860
Researchers: Thomas Dannenhoffer-Lafage, Jaehyeok Jin, Yining Han
The Theory of Ultra-Coarse-Graining (UCG)
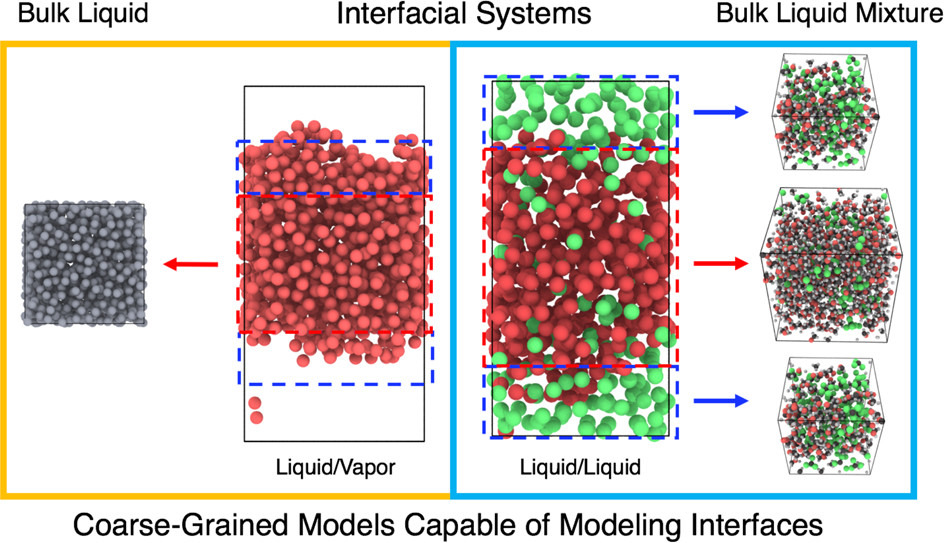
We developed a new systematic framework for coarse-graining by incorporating internal state variables with configurational variables into CG beads. This UCG he development of this method is actively ongoing. In the current UCG framework, two different internal dynamics are rigorously explored: rapidly equilibrating (in a local equilibrium) or rarely switching (Markov-state). For the rarely switching dynamics, we constructed the UCG model at ultralow resolution that combines particle-based and Markov-state-based modeling to allow for dynamics both of and within coarse-grained beads. From the developed UCG model, we studied the limit where state switching is rare compared to the time scale of particle motion.

For the other extreme where the internal CG states are in a rapid local equilibrium, the UCG force field is obtained by mixing standard CG force fields in terms of local order parameters where the degree of mixing is regulated. We designed the UCG theory for this limit to embed an environmental dependence and many-body effects to the CG force field. To modulate the interactions between the UCG beads, we focused on order parameters in various systems ranging from hydrophobic association to inhomogeneous interfacial systems. Compared to the standard CG approach, the developed UCG models can faithfully recapitulate the structural correlations more accurately. More fascinatingly, we recently demonstrated that this UCG approach can impart transferable CG interactions from the bulk system to the interfacial systems (liquid-vapor or liquid-liquid interfaces). Currently, we are working on applying the UCG methodology to complex biomolecules or liquid phenomena.
Relevant Papers:
-
Jin, J., Voth, G. A. (2018). Ultra-Coarse-Grained Models Allow for an Accurate and Transferable Treatment of Interfacial Systems. Journal of Chemical Theory and Computation, 14(4), 2180-2197. doi: 10.1021/acs.jctc.7b01173
-
Dama, J. F., Jin, J., Voth, G. A. (2017). The Theory of Ultra-Coarse-Graining. 3. Coarse-Grained Sites with Rapid Local Equilibrium of Internal States. Journal of Chemical Theory and Computation, 13(3), 1010-1022. doi: 10.1021/acs.jctc.6b01081
-
Davtyan, A., Dama, J. F., Sinitskiy, A. V., Voth, G. A. (2014) The Theory of Ultra-Coarse-Graining. 2. Numerical Implementation. Journal of Chemical Theory and Computation, 10(12), 5265–5275. doi: 10.1021/ct500834t
-
Dama, J. F., Sinitskiy, A. V., McCullagh, M., Weare, J., Roux, B., Dinner, A. V., Voth, G. A. (2013). The Theory of Ultra-Coarse-Graining. 1. General Principles. Journal of Chemical Theory and Computation, 9(5), 2466–2480. doi: 10.1021/ct4000444
Researchers: Jaehyeok Jin
CG Dynamics
Due to the dramatic reduction in the number of degrees of freedom (DoF) that is achieved through coarse-graining, the dynamics in the CG system is usually sped up. However, the interpretation of CG dynamics is often difficult due to the inhomogeneous acceleration of diffusion and transitions. This is due to the removal of the fluctuation forces on the CG sites after the DoF reduction. Our dynamic force matching method allows for the reintroduction of those fluctuation forces into the CG model through “fictitious particles” in a way that does not significantly affect the computational efficiency or the equilibrium properties of the CG model. In our first paper, we showed that this method can accurately reproduce the diffusion rate and reasonably represent the short-time dynamics. Additional developments have allowed us to simultaneously reproduce multiple long-time properties (such as self-diffusion rate and rotational relaxation rate) as well.
Relevant Papers:
-
Davtyan, A., Voth, G. A., Andersen, H. C. (2016) Dynamic Force Matching: Construction of Dynamic Coarse-Grained Models with Realistic Short Time Dynamics and Accurate Long Time Dynamics. Journal of Chemical Physics. 145, 224107. doi: 10.1063/1.4971430
- Davtyan, A., Dama, J. F., Voth, G. A., Andersen, H. C. (2015). Dynamic force matching: A method for constructing dynamical coarse-grained models with realistic time dependence. Journal of Chemical Physics. 142, 154104. doi: 10.1063/1.4917454
Researchers: Yining Han, Jaehyeok Jin
Enhanced Sampling Development
Creating coarse-grained models that properly reflect the underlying physics and chemistry inherently requires having robustly explored the phase space defined by the fine-grained model. Moreover, it is important that the model be parameterized correctly in order to have simulations properly reflect behavior seen in experiments. The Voth group has sought to ameliorate difficulties related to both aspects of the sampling problem.
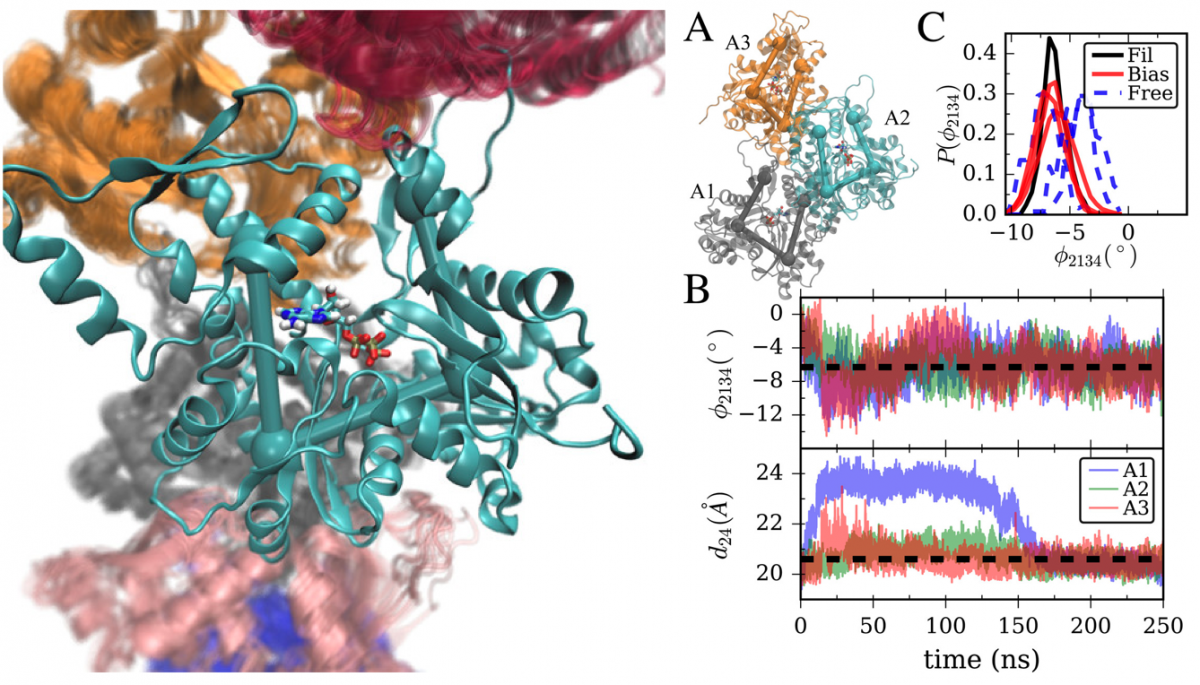
First, in order to ensure robust sampling, the Voth group makes extensive use of free energy methods, which enhance the exploration of phase space along chosen collective variables (reaction coordinates) in the system. We make extensive use of both umbrella sampling and metadynamics. We have recently put a substantial amount of effort into the development and benchmarking of new metadynamics variants. In particular, our group first proved that the well-tempered version of metadynamics converges asymptotically to the exact free energy of the system. Then using the conditions for convergence, we were able to design new methods, such as the transition tempered metadynamics method, which first uses untempered metadynamics to enhance exploration, then tempers after a transition between two specified areas of phase space is detected, giving much more robust results quickly. For example, we have shown this to be a superior method for studying the permeation of small molecules through lipid bilayers. We have also recently developed the metabasin metadynamics method, which restricts the area in which bias is added to a region which is below a free-energetic threshold, where this region is detected automatically and on-the-fly. This allows for more careful addition of bias without pushing the system into unphysical regions.

Second, in order to ensure the distribution sampled reflects experimental observations, new linear biasing methods have been developed that ensure experimentally observed averages or target distributions are recovered by a simulation with minimal perturbation. We have developed experimentally directed simulation (EDS) methods which discover the correct biasing parameters on the fly, and show how these can also be used to bias coarse-grained models. We have also developed an experimentally directed metadynamics method (EDM), which uses the same ideas of metadynamics to add bias to the system such that the system samples a target distribution. The EDS method has recently been applied in reactive simulations of water and water with an excess proton. Here we show that by only correcting the oxygen-oxygen radial distribution function of DFT water with a poor functional, we can greatly improve the structure and dynamics in the system effectively without additional cost.
All of these new methods are all available and implemented in public repositories:
Transition tempered and metabasin: https://github.com/JFDama/plumed2/tree/pinion-cleanup
Relevant Metadynamics Papers:
-
Sun, R., Han, Y., Swanson, J. M., Tan, J. S., Rose, J. P., Voth, G. A. (2018). Molecular Transport Through Membranes: Accurate Permeability Coefficients From Multidimensional Potentials of Mean Force and Local Diffusion Constants. Journal of Chemical Physics, 149, 072310. doi: 10.1063/1.5027004
-
Sun, R., Dama, J. F., Tan, J. S., Rose, J. P., Voth, G.A. (2016). Transition-Tempered Metadynamics is a Promising Tool for Studying the Permeation of Drug-like Molecules through Membranes. Journal of Chemical Theory and Computation. 12(10), 5157–5169. doi: 10.1021/acs.jctc.6b00206
-
Dama, J. F., Hocky, G. M., Sun, R., Voth, G. A. (2015). Exploring Valleys Without Climbing Every Peak: More Efficient and Forgiving Metabasin Metadynamics via Robust On-the-Fly Bias Domain Restriction. Journal of Chemical Theory and Computation. 11(12), 5638–5650. doi: 10.1021/acs.jctc.5b00907
-
Dama, J. F., Rotskoff, J., Parrinello, M., Voth, G.A. (2014). Transition-Tempered Metadynamics: Robust, Convergent Metadynamics via On-The-Fly Transition Barrier Estimation. Journal of Chemical Theory and Computation. 10(9), 3626–3633. doi: 10.1021/ct500441q
-
Dama, J. F., Parrinello, M., Voth, G. A. (2014). Well-tempered Metadynamics Converges Asymptotically. Physical Review Letters. 112, 1-6. doi: 10.1103/PhysRevLett.112.240602
Relevant Experiment Directed Papers:
-
Hocky, G. M., Dannenhoffer-Lafage, T., Voth, G. A. (2017). Coarse-Grained Directed Simulation. Journal of Chemical Theory and Computation, 13(9), 4593-4603. doi: 10.1021/acs.jctc.7b00690
-
White, A. D., Knight, C., Hocky, G. M., Voth, G.A. (2017). Improved Ab Initio Molecular Dynamics by Minimally Biasing with Experimental Data. Journal of Chemical Physics. 146, 041102. doi: 10.1063/1.4974837
-
Dannenhoffer-Lafage, T., White, A. D., Voth, G. A. (2016). A Direct Method for Incorporating Experimental Data into Multiscale Coarse-grained Models. Journal of Chemical Theory and Computation. 12(5), 2144-2153. doi: 10.1021/acs.jctc.6b00043
-
White, A. D., Dama, J. F., Voth, G. A. (2015). Designing Free Energy Surfaces that Match Experimental Data with Metadynamics. Journal of Chemical Theory and Computation. 11(6), 2451-2560. doi: 10.1021/acs.jctc.5b00178
- White, A. D., Voth, G. A. (2014). Efficient and Minimal Method to Bias Molecular Simulations with Experimental Data. Journal of Chemical Theory and Computation. 10(8): 3023–3030. doi: 10.1021/ct500320c
Researchers: Thomas Dannenhoffer-Lafage, Glen Hocky